
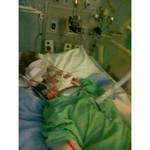
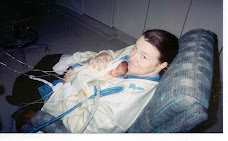
On Saturday I got a sweet letter from the metabolic geneticist outlining the tests they did in the hospital before Hannah passed away and the results. Everything came back normal. I really wasn't surprised. I think we mostly just had them done to rule out syndromes that are common in kids who have seizures.
This is what I believe Hannah had (just needed a little more time to get to Chicago. I got wrapped up in the "hows" of getting her there etc. and just thought we had time. :( Her epi here wanted to rule out everything else before considering Dravet.)
Dravet syndrome
INTRODUCTION
Dravet (Dra-vay) syndrome, previously known as Severe Myoclonic Epilepsy of Infancy (SMEI), is a neurodevelopmental disorder beginning in infancy and characterized by severe epilepsy that does not respond well to treatment. Estimates of the prevalence of this rare disorder have ranged from 1:20,000 to 1:40,000 births, though the incidence may be found to be greater as the syndrome becomes better recognized and new genetic evidence is discovered. It is thought to occur with similar frequency in both genders and knows no geographic or ethnic boundaries.
The course of Dravet syndrome is highly variable from person to person. Seizures begin during the first year of life and development is normal prior to their onset. In most cases, the first seizures occur with fever and are generalized tonic-clonic (grand mal) or unilateral (one-sided) convulsions. These seizures are often prolonged and may lead to status epilepticus, a medical emergency. In time, seizures increase in frequency and begin to occur without fever. Additional seizure types appear, most often these are myoclonic, atypical absence, and complex-partial seizures.
During the second to fourth year of life, varying degrees of developmental delay typically become apparent and can include regression of aquired skills.
Additional features that are seen in significant numbers of patients with Dravet syndrome may include sensory integration disorders and other autism spectrum characteristics, orthopedic or movement disorders, frequent or chronic upper respiratory and ear infections, sleep disturbance, dysautonomia, and problems with growth and nutrition.
GENETICS
The sodium channel gene SCN1A is currently the most clinically relevant gene found to cause epilepsy and is a known contributor to a significant percentage (approximately 50-80%) of cases of Dravet syndrome. Researchers have documented many different mutations of the SCN1A gene, however the majority of them do not result in Dravet syndrome. SCN1A mutations most often lead to milder forms of epilepsy, including Generalized Epilepsy with Febrile Seizures (GEFS), Generalized Epilepsy with Febrile Seizures Plus (GEFS+), Intractable Childhood Epilepsy with Generalized Tonic-Clonic Seizures (ICEGTC), and Severe Myoclonic Epilepsy, Borderline (SMEB). Dravet syndrome is generally considered to be the severe end of a broad spectrum of SCN1A-related epilepsies.
GEFS -------------- GEFS+ ----------- ICEGTC ------------ SMEB -------------- SMEI/Dravet syndrome
Mutations of the SCN1A gene have an autosomal dominant inheritance pattern, meaning that they can be passed from parent to child. However, in Dravet syndrome, although at least one-fourth of the affected individuals have some history of febrile seizures or epilepsy in their extended family, the gene mutation nearly always arises “de novo”, or new to the individual, meaning that neither parent tests positive for the gene mutation and it is thought to have occurred spontaneously after conception. Modifying genes and environmental factors likely play an important role in determining the severity of the resulting condition. Other genes have also recently been implicated in Dravet syndrome, including SCN9A and PCDH19. Much remains to be understood about the causes of Dravet syndrome and research is ongoing. Although Hannah tested normal for SCN1A and PCDH19 I have friends who have had their children retested and the mutation was found the second time. Also you can be clinical for Dravet without the gene mutation.
TREATMENT
At this time, the treatments available for Dravet syndrome are focused on improving symptoms, primarily anticonvulsant medications to control seizures. Response to these medicines is variable, but often seizures persist despite treatment. While certain medications have been found to be generally useful for individuals with Dravet syndrome, others have been quite consistently found to have an aggravating effect. The helpfulness of other anticonvulsant therapies, such as the Ketogenic Diet and vagus nerve stimulation (VNS), are in ongoing evaluation. Once again, results tend to be highly variable from person to person.
Early implementation of global therapies is essential to support optimal development. Patients with Dravet syndrome should receive physical, occupational, speech, and social/play therapies and an enriched environment is encouraged. Treatments to address orthopedic, sleep, autonomic, immune and growth problems may be important considerations.
OUTCOMES
Outcomes, once again, tend to be variable. For many individuals, the progression of Dravet syndrome begins to stabilize after the age of four. Partial and myoclonic seizures may attenuate, and in some cases disappear. Convulsive seizures, though their frequency and intensity may moderate, usually persist, often occurring during sleep. Fever continues to provoke seizures and can still lead to status epilepticus. Communication, motor, and cognitive function stabilize, but significant delays remain to varying degrees. Despite being at increased risk for accidents, infection, status epilepticus, and Sudden Unexplained Death in Epilepsy (SUDEP), an individual with Dravet syndrome has an 85% chance of surviving into adulthood. Because this disorder is rare and has relatively recently been identified as a distinct syndrome, little is known about long-term prognosis and life expectancy.
http://www.idea-league.org/dravet-syndrome You can read more extensively and follow links at this site.
I posted this information for any other moms who may be following Hannah's story and they need this information. I only learned about Dravet a couple years ago and I learned about it from parents who had kids with it. I wish I had been more "pushy". Not that it would have changed the outcome but for my own peace of mind. Please support the Idea League and what they are doing to help these kiddos. Through them I have learned of many other children like Hannah who have left their families way too soon. It's a devastating experience and one that hopefully some day is preventable.
Monday, March 15, 2010
Subscribe to:
Post Comments (Atom)
















3 comments:
I know of two families who have lost children to Dravets. It is a terrible form of epilepsy (as if any of them are any good!) and I am so so sorry to read about Hannah. I have two daughters, the oldest is almost 17 and she has had epilepsy since she was 13 months. I have met many wonderful amazing parents and children during that time that I never would have otherwise met.
I don't know if this will help you at all but a wonderful mom and dad (Sara and David) whose little girl, Daisy, had Dravets and died 6 years ago have started a foundation in her memory. The ketogenic diet was the only thing that halped Daisy's seizures so the main focus of their foundation is funding dieticians for the keto diet in the UK (where they live) because it is not well known there.
Anyway I thought you might like to check out their webite, maybe even contact them. Sara is a lovely, lovely woman.
http://www.thedaisygarland.org.uk/
Again, words are useless but I am so sorry for what you have been through.
Epilepsy is a horrible disease that attacks many people in the world ... I Ustaria know more about epilepsy but also thank you very much for your post!
This information is very interesting, I really enjoyed, I would like get more information about this, because is very
beautiful, thanks for sharing! costa rica investment opportunities
Post a Comment